16.7 Evolución de los métodos de secuenciación masiva
Los métodos de secuenciación de Maxam y Gilbert y de Sanger (pág. 248) —ambos publicados en 1977— supusieron una revolución en la investigación del material genético. En particular, las mejoras técnicas del método enzimático de Sanger en años posteriores fueron la clave para la consecución de los objetivos del Proyecto Genoma Humano en 2001.
La evolución del método original de Sanger puede resumirse en
- utilización de nucleótidos terminadores marcados con 4 fluorocromos, en lugar del isótopo radiactivo original;
- detección mediante láser de los 4 fluorocromos simultáneamente en una misma muestra electroforética;
- utlización de la electroforesis capilar para reducir la cantidad de muestra necesaria, para aumentar la velocidad y resolución de la separación, así como para permitir el procesado simultáneo de decenas de muestras (típicamente, 96).
Con tales avances y la automatización e informatización asociadas se consiguió aumentar el ritmo de secuenciación de un instrumento desde alrededor de 25 kb por semana en 1990 hasta unos miles de kb en 2001.
A partir de 2005 han ido apareciendo diversas técnicas que se basan en principios completamente diferentes a la de Sanger y permiten obtener secuencias con una rapidez varios órdenes de magnitud superior. Además, reducen sustancialmente el coste económico del proceso por cada base secuenciada. En ellas, la incorporación de matrices bidimensionales de detectores ópticos (del tipo cámaras CCD o matrices de diodos) ha permitido elevar la cifra hasta cerca de 109 canales de análisis simultáneo (es decir, ese número de muestras se analiza en paralelo en cada experimento). Con ello se definen las nuevas metodologías de secuenciación como masivamente paralelas. Se reduce, sin embargo, el número de bases consecutivas que pueden secuenciarse en un experimento, muy por debajo de las 600-1200 típicas de la tecnología anterior. Aun con esto, la cifra semanal se eleva hasta 250 gigabases secuenciadas por un solo instrumento.
Todas estas técnicas comprenden tres grandes fases: la preparación de las plantillas (moldes para la síntesis de una hebra nueva de DNA), la secuenciación y captación de imagen (señal), y el análisis de datos. No se comentará aquí nada sobre la tercera, por ser más técnica y especializada.
Las diferencias con la tecnología tradicional pueden resumirse en cuatro aspectos principales:
- La preparación de las muestras es más breve y sencilla, y no utiliza clonación y purificación de los DNA.
- Los fragmentos se amplifican sobre un soporte sólido (una superficie o bien microesferas en suspensión).
- Los fragmentos amplificados se fijan sobre una superficie de vidrio, un biochip (pág. 170) o microesferas, o se atrapan en una capa delgada de gel de poliacrilamida, o bien se depositan en los micropocillos de una placa especialmente diseñada.
- Sobre ese soporte se llevan a cabo las reacciones de secuenciación repetitivas. Dichas reacciones son diferentes en las metodologías o plataformas que distintas empresas han desarrollado paralelamente, pero todas ellas tienen lugar de forma escalonada, un nucleótido en cada etapa (en contraste con la secuenciación tradicional, que separa las mezclas finales resultantes de numerosas etapas de reacción sucesivas).
Preparación de las plantillas
Como se ha indicado, la muestra de DNA se fragmenta de forma aleatoria por medios enzimáticos o mecánicos, rindiendo una población de moléculas de tamaño reducido (<1 kb, por ejemplo). A la colección o biblioteca de fragmentos resultante se ligan adaptadores (pág. 207) sobre sus extremos. Los adaptadores, olignucleótidos sintéticos comunes a todos, permiten amplificar todos los fragmentos con una misma pareja de cebadores.
A continuación se efectúa la amplificación de las moléculas que se han distribuido previamente de forma separada en el espacio, de modo que se generen poblaciones discretas de moléculas idénticas. El conjunto cubre todo el DNA de la muestra original. Se emplea una polimerasa y técnicas derivadas de la PCR. Esta amplificación es necesaria porque la mayoría de sistemas no pueden detectar la emisión de un solo cuanto de luz (un evento fluorescente o luminiscente producido por una sola molécula).
Finalmente, las moléculas de DNA se fijan a un soporte sólido —si no se ha hecho ya en la etapa previa. Pueden seguirse tres abordajes alternativos, dependiendo de la metodología:
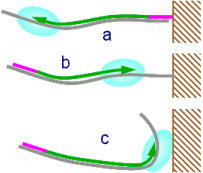
- Pueden inmovilizarse los cebadores que se utilizarán para la reacción de secuenciación. A ellos se unen las moléculas de la muestra, ejerciendo de molde para la elongación de los cebadores.
- Pueden fijarse las propias moléculas de la muestra amplificada. A continuación, los cebadores se unen a ellas y se elongan usándolas como molde.
- En algunas tecnologías se fijan al soporte moléculas de polimerasa, que unirán cada molécula molde de DNA de la muestra y sintetizarán su complemtaria a partir del cebador emparejado al molde.
Reacciones de secuenciación y detección
En esta fase hay mayores diferencias entre las diversas plataformas que se han desarrollado. Las que están en uso actualmente se pueden clasificar así:
Terminación reversible cíclica
En este método se usa una DNA polimerasa, que utiliza el DNA amplificado de la muestra como molde y el cebador como iniciador. Se basa en la adición a la cadena creciente de un nucleótido terminador —es decir, la polimerasa lo reconoce y lo une a la cadena pero no admite una subsiguiente adición. Este planteamiento es similar al método de Sanger, salvo que en este caso el bloqueo puede eliminarse antes de la siguiente ronda para que la elongación continúe. Es decir, en cada etapa se permite la adición de un único nucleótido, que se detecta de inmediato, antes de desbloquearlo para pasar a la etapa siguiente.
Los nucleótidos especiales (terminadores reversibles) que se añaden se incorporan a la cadena nueva y se detectan gracias a que llevan un fluoróforo unido. Si se usan fluorocromos de 4 colores pueden añadirse los 4 tipos de nucleótido a la vez; en caso contrario, se añade uno solo por etapa. Para poder pasar a la siguiente etapa es preciso escindir tanto el grupo protector como el fluoróforo.
Existen dos tipos de terminadores:
- Nucleótidos con un grupo protector en su OH 3' y un fluoróforo unido a la base nitrogenada. Tras la detección de fluorescencia, se deben eliminar ambos mediante reacciones específicas.
Ejemplos
Ejemplos de terminadores reversibles bloqueados:
grupo que bloquea el OH 3' (alilo, azidometilo);
posición donde va unido el fluoróforo;
enlace que se puede escindir;
parte adicional que queda unida al nucleótido tras la escisión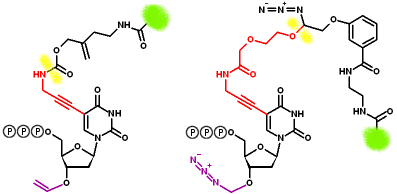 (Pulse para cerrar este cuadro)
(Pulse para cerrar este cuadro) - Nucleótidos terminadores "no bloqueados". El sustituyente unido a la base (que incluye el fluoróforo) evita, por impedimento estérico u otro mecanismo, que se incorpore el siguiente nucleótido. Tras la detección sólo es preciso escindir un enlace.
Ejemplos
Ejemplos de terminadores reversibles bloqueados:
grupo que impide la elongación;
posición donde va unido el fluoróforo;
enlace que se puede escindir;
parte adicional que queda unida al nucleótido tras la escisión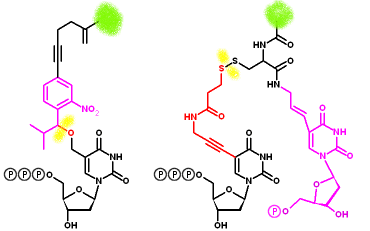 (Pulse para cerrar este cuadro)
(Pulse para cerrar este cuadro)
Adición de un solo nucleótido: pirosecuenciación
Se utiliza igualmente una polimerasa para sintetizar una hebra complementaria a la muestra, pero no se marcan los nucleótidos. En cambio, se detecta la liberación de una molécula de pirofosfato propia de cada reacción de elongación. El PPi inicia una serie de reacciones acopladas que termina con la emisión de luz detectable.
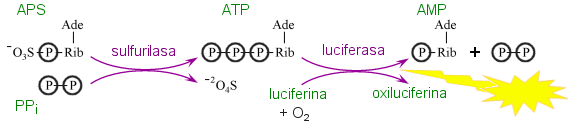
Al añadir un solo tipo de nucleótido en cada etapa, y en cantidad limitante, cada señal positiva indica que se ha incorporado y, por tanto, se sabe cuál es el que hay en la hebra molde fijada al soporte sólido.
Ligamiento dirigido por el molde
En este caso no se utiliza polimerasa, sino una DNA ligasa. Se añaden sucesivamente oligonucleótidos de secuencia conocida y marcados con fluorocromo. Si emparejan con la hebra molde en posición adyacente al cebador, la ligasa los une a éste y se detecta la fluorescencia. A continuación, mediante una hidrólisis química específica se libera el extremo 3' del oligonucleótido añadido, junto con su fluorocromo. Repitiendo el proceso numerosas veces en cada etapa, con oligonucleótidos de secuencias diversas pero conocidas, es posible reconstruir la secuencia completa del molde.
La descripción anterior está muy simplificada.
Más concretamente, los oligonucleótidos que se utilizan se denominan sondas "codificadas" en una o dos bases. Esto significa que hay una o dos bases cuya identidad se varía de modo controlado —en combinación con diferentes fluorocromos— y sirve para analizar o "interrogar" la secuencia desconocida (si emparejan con el molde, se sabe qué base tiene éste en esa posición). En el resto del posiciones del oligonucleótido se cubren todas las posibilidades, bien empleando una mezcla de todas las secuencias posibles o bien utilizando bases que emparejen con cualquier base en el molde; éstas se conocen como bases degeneradas cuando una misma base puede emparejarse con 2 o 3 diferentes (algo similar a lo que ocurre en el balanceo del anticodón, pág. 315), y bases universales cuando se integran en la doble hélice sin formar enlaces de hidrógeno y, por tanto, pueden enfrentarse a cualquier base.
Una vez el oligonucleótido sonda se ha ligado, se escinde su parte 3', lo que incluye el fluoróforo y la mayoría de los nucleótidos degenerados o universales, para proceder a la adición de una nueva sonda que se pueda ligar a continuación de la precedente.
Secuenciación en tiempo real
El nombre indica que se detecta la incorporación de forma continua, sin pausas. Concretamente, en una de las formulaciones se detectan los nucleótidos mientras están asociados a la polimerasa, que está adsorbida sobre una superficie. Se utiliza una tecnología de detección (detector de guía de onda de modo cero) que sólo es sensible a la emisión de fluorescencia en contacto íntimo con la superficie.
Suelene utilizarse como sustratos para la polimerasa nucleósidos-polifosfato con el fluoróforo unido al último grupo fosfato. Una vez que el NMP se añade a la nueva hebra, el fluoróforo queda separado y se elimina antes de la siguiente etapa. Ejemplos
enlace que se escinde en la incorporación;
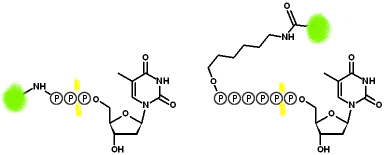
Secuenciación de molécula individual
Las metodologías de desarrollo más reciente están abordando la secuenciación de una sola molécula de DNA, sin amplificación. Sin entrar en detalles, la principal ventaja de esta aproximación es que se evita la introducción de errores de replicación durante la PCR, que en los otros métodos hace que la población clonal de moléculas que se secuencian tenga microheterogeneidad.