Cómo se hizo
el simulador de espectros de dicroísmo circular de proteínas
Datos de partida
Espectros canónicos de hélice alfa, hebra beta, giro y aleatoria.
El dato de partida fue el archivo de imagen ss_960531_AFrame_62 de PPS'96 (Principles of Protein Structure Using the Internet) (disponible en varios sitios; por ej.:
- The Principles of Protein Structure '97
http://www.med.unibs.it/~marchesi/pps97/course/section8/ss-960531_21.html#HEADING25
- http://www.med.unibs.it/~marchesi/pps97/course/section8/ss_960531_AFrame_62.gif
- The Principles of Protein Structure '97
http://www.cryst.bbk.ac.uk/pps97/course/section8/ss-960531_21.html#HEADING25
- http://www.cryst.bbk.ac.uk/pps97/course/section8/ss_960531_AFrame_62.gif
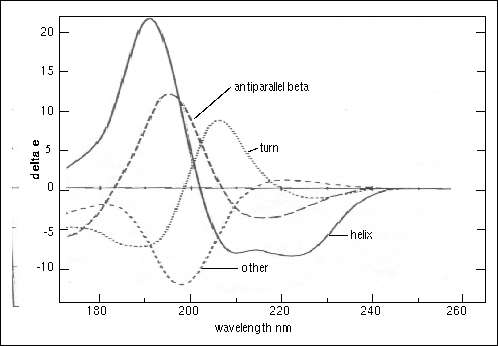
Figure 23.
Circular dichroism spectra of "pure" secondary structures. Redrawn from Brahms & Brahms, 1980.
Brahms, S. & Brahms, J. (1980) Determination of protein secondary structure in solution by vacuum ultraviolet circular dichroism J. Mol. Biol. 138, 149-178.
Digitalizado semimanualmente con Engauge Digitizer (de SourceForge, http://digitizer.sourceforge.net/) [De acuerdo con el historial de versiones en SF, debió de ser la v. 2.12]
Generó archivos con datos (X,Y), en formato delimitado por comas (CSV).
Suavizados en SigmaPlot usando Transforms > Smooth 2D data >
- Smoother = Negative exponential
- Sampling proportion = 0.2
- Polynomial degree = 4
Intenté ajustar a 3 gaussianas; alfa y beta ajustaban bastante bien,
pero no lo usé.
Con la función interpolar generó sendos juegos de datos con una X común y espaciada regularmente:
- interv=data(170,255,1)
- col(15)=interv
- col(N)=interpolate(col(A),col(B),interv)
- donde N=16 a 19, A=1 a 7, B=2 a 8
Superponen my bien con los espectros de ejemplo,
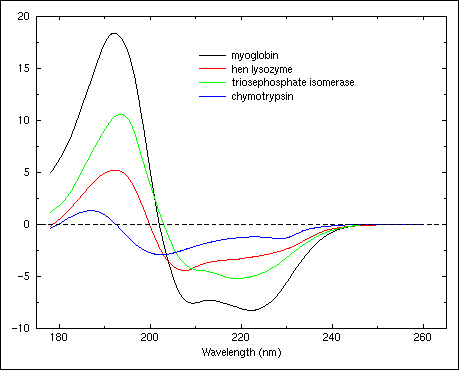
Figure 22.
Circular dichroism spectra of triosephosphate isomerase (H:0.52, S:0.14, T:0.11, O:0.23), hen egg lysozyme (H:0.36, S:0.09, T:0.32, O:0.23), myoglobin (H:0.78, S:0.0, T:0.12, O:0.10), and chymotrypsin (H:0.10, S:0.34, T:0.20, O:0.36).
Con ellos puedo crear una función generadora de espectros combinados.
Implementación en página web:
 Una primera versión se basó en el método gráfico de Jeffrey Clymer (bastante reescrito):
Una primera versión se basó en el método gráfico de Jeffrey Clymer (bastante reescrito):
- http://members.nuvox.net/~on.jwclymer/graphs/plot.html ==> Please go to: http://home.cincyarts.org
- Graphing in JavaScript. Plots functions of your choosing.
http://home.fuse.net/clymer/graphs/plot.html
[ jwclymer@one.net Debut: December 26, 2003. Revision No. 3. Wednesday, December 29, 2004 ]
Esto funcionaba bien, pero producía líneas punteadas.
 Por ello, se adoptó en su lugar el empleo de la biblioteca gráfica en JavaScript de Walter Zorn para conseguir el dibujo de la gráfica.
Por ello, se adoptó en su lugar el empleo de la biblioteca gráfica en JavaScript de Walter Zorn para conseguir el dibujo de la gráfica.
- http://www.walterzorn.com/jsgraphics/jsgraphics_e.htm
Los datos empleados para construir las gráficas son:
- los espectros canónicos digitalizados (listas de valores X,Y) se exportaron a formato CSV y se convirtieron en matrices de datos JavaScript, que se usan en la página para calcular una serie de valores Y (píxeles en pantalla) para cada X (píxeles en pantalla);
- para generar cualquier combinación de estructuras secundarias, se hace una combinación lineal de los espectros canónicos ponderada con la fracción de cada tipo de estructura secundaria;
- para las proteínas de ejemplo, los datos del pie de la fig.22 de PPS (arriba);
- todo ello usando JavaScript.
Angel Herráez. Parte de la sede web Biomodel.uah.es