La mayoría de las biomoléculas poseen una carga eléctrica, cuya magnitud depende del pH del medio en el que se encuentran. Como consecuencia, se desplazan cuando se ven sometidas a un campo eléctrico.
Se denomina electroforesis a la técnica mediante la cual se separan las biomoléculas en disolución cuando se ven sometidas a un campo eléctrico. Se trata de una técnica fundamentalmente analítica, aunque también se puede realizar con fines preparativos.
Cada molécula se desplaza por efecto del campo, alcanzando rápidamente una velocidad constante al equilibrarse la fuerza impulsora (fuerza del campo eléctrico) con la resistencia al avance (fuerza de fricción o rozamiento) impuesta por el medio en el que se desplaza.
| Fuerza del campo eléctrico | = | Fuerza de fricción |
| `q * E` | `=` | `f*v` |
| `q` = carga (C) `E` = intensidad del campo (V/m = N/C) |
`f` = coeficiente de fricción (C·V·s / m2 = kg/s) `v` = velocidad de la molécula (m/s) |
| Fuerza del campo eléctrico | = | Fuerza de fricción |
| q × E | = | ƒ × v |
| q = carga (C) E = intensidad del campo (V/m = N/C) |
ƒ = coeficiente de fricción (C·V·s / m2 = kg/s) v = velocidad de la molécula (m/s) |
El coeficiente de fricción mide la resistencia intrínseca debida a las características de cada molécula, siendo éstas esencialmente su forma y su tamaño. Así, por ejemplo, las moléculas grandes y de forma irregular poseen un mayor coeficiente de fricción que las pequeñas y compactas.
La velocidad por unidad de campo recibe el nombre de movilidad electroforética, μ
`v/E = mu = q/(f) = (Ze)/f`
(`Z` = número entero; `e` = carga del electrón)
v / E = μ = q / ƒ = Ze / ƒ
(Z= número entero; e = carga del electrón)
En unas condiciones determinadas de electroforesis, la diferente movilidad de cada molécula definirá su separación en el espacio; al ir transcurriendo el tiempo, se van separando progresivamente unas de otras.
Al ser el medio de soporte a su vez un polielectrolito, está constituido por iones, que son atraídos y así recubren los iones de la muestra, lo que altera el comportamiento de éstos en el campo. Todo ello hace que el tratamiento teórico cuantitativo de la electroforesis sea muy difícil y que esta técnica resulte poco útil para obtener información precisa sobre la estructura de las moléculas. Sin embargo, es enormemente útil como técnica analítica y preparativa.
Se suele hablar de 3 tipos de electroforesis:
Estudiaremos únicamente la electroforesis zonal, de uso más extendido.
Finalmente, debe señalarse que, aunque es menos frecuente, también se puede aplicar la electroforesis para la separación de células.
La muestra debe situarse en o sobre un medio soporte, principalmente para evitar perturbaciones mecánicas y corrientes de convección durante la separación. En algunos soportes (papel o similares) la muestra queda sobre la superficie y avanza a lo largo de ella, con escasa fricción, por lo que el mecanismo principal de separación es la magnitud de la carga de cada componente de la muestra. Otros medios de soporte (“geles”, medios semisólidos o gelatinosos) están formados por polímeros que forman una malla, matriz o red tridimensional a través de la cual deben avanzar las moléculas de la muestra, que queda embebida en el medio de soporte electroforético. Como consecuencia, la fricción es notable y los factores de forma y tamaño adquieren una alta relevancia en la separación.
| Medios de baja fricción | Medios de elevada fricción | |
| papel acetato de celulosa |
gel de almidón gel de agarosa |
gel de poliacrilamida gel de agarosa+poliacrilamida |
| separación principalmente por carga | separación por carga, tamaño y forma | |
Papel: sencillo, pero con elevada adsorción debido a los grupos hidroxilo de la celulosa.
Acetato de celulosa: los grupos –OH están acetilados, lo que reduce la adsorción; baja tinción de fondo; es posible transparentarla o disolverla para detectar y recuperar, respectivamente, los componentes separados.
Almidón: pasta de almidón cuyos granos se han disgregado en un tampón caliente (se hinchan). Actualmente se utiliza poco, ha sido sustituido por la poliacrilamida.
Agarosa: polisacárido, producto purificado de algas (composición similar al agar-agar). Se disuelve en caliente (50-60°C) y al enfriar solidifica formando un gel, de alta porosidad.
Poliacrilamida: el gel es el resultado de la polimerización química de una mezcla de acrilamida y bisacrilamida. Regulando la concentración de ambas y su proporción se consiguen distintas porosidades, siempre menores que la de los geles de agarosa.
| acrilamida: | 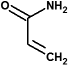 |
(forma polímeros lineales) |
| N,N’-metileno-bis(acrilamida): | 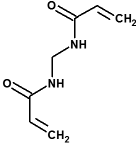 |
(introduce uniones cruzadas entre cadenas de poliacrilamida) |
| poliacrilamida: (vista parcial) |
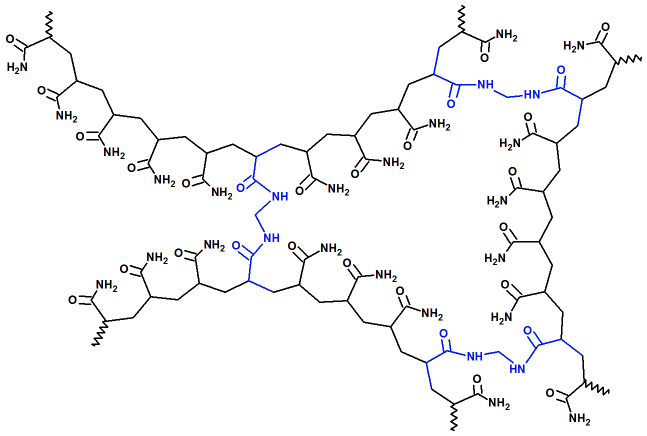 |
|
Agarosa+poliacrilamida: se consigue una porosidad intermedia.
Poliacrilamida con SDS: el dodecilsulfato sódico (Sodium Dodecyl Sulphate; sinónimo: laurilsulfato sódico) es un detergente aniónico que se une a las proteínas, desnaturalizándolas en una conformación extendida recubierta de moléculas de SDS. Como consecuencia, el tamaño de la molécula de proteína es directamente proporcional a su longitud en aminoácidos y su carga queda enmascarada por la mayor carga del SDS que la recubre, que es también proporcional a la longitud. Por lo tanto, la movilidad electroforética de la proteína depende exclusivamente de su masa molecular.
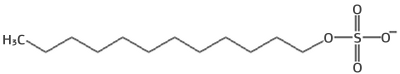 H-(CH2)12-SO4−
H-(CH2)12-SO4−
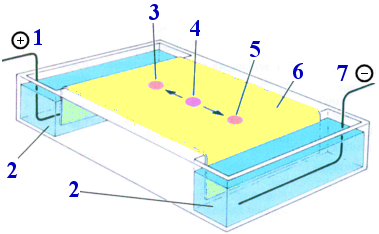 |
1. ánodo (+) 2. compartimentos con tampón y los electrodos 3. componente que migra debido a su carga negativa 4. punto de aplicación de la muestra 5. componente que migra debido a su carga positiva 6. papel 7. cátodo (−) |
En papel, para aminoácidos u otras moléculas pequeñas; en soportes similares (especialmente, acetato de celulosa), para proteínas.
En gel de almidón o de agarosa, para proteínas y especialmente para ácidos nucleicos. Casi siempre el tampón cubre el gel (para evitar que se seque debido al calentamiento sufrido al pasar la corriente), denominándose por ello “electroforesis submarina”.
El soporte se impregna por capilaridad de disolución tampón, que disuelve la muestra y mantiene el contacto eléctrico.
Se aplica la muestra depositándola (con pipeta o un aplicador específico) como una gota sobre el soporte (papel, acetato de celulosa) o dentro de un “pocillo” creado en el gel.
Se usa casi exclusivamente con gel de poliacrilamida (más resistente físicamente, no se desliza), para proteínas o para ácidos nucleicos de pequeño tamaño.
El gel puede rellenar tubos de vidrio (este formato ya apenas se usa) o estar contenido entre 2 placas rectangulares.
Contacto eléctrico y disolvente gracias al tampón que embebe el gel y llena las cubetas o compartimentos del ánodo y cátodo.
La muestra se deposita con micropipeta llenando un “pocillo” creado al polimerizar el gel.
En cuanto a la composición del gel hay dos variantes: electroforesis continua (un solo tipo de gel) y electroforesis discontinua (2 tramos de gel de composición ligeramente diferente).
1. compartimento superior con tampón y el cátodo 2. bandas en las que se separan los componentes 3. colorante indicador del frente de avance 4. compartimento inferior con tampón y el ánodo |
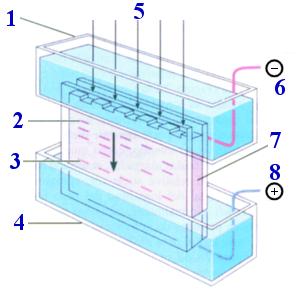 |
5. pocillos donde se aplican las muestras 6. cátodo (−) 7. gel colocado entre placas de vidrio 8. ánodo (+) |
1. compartimento superior con tampón y el cátodo 2. gel colocado entre placas de vidrio 3. compartimento inferior con tampón y el ánodo |
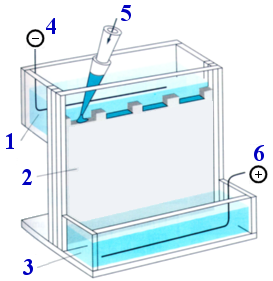 |
4. cátodo (−) 5. las muestras se aplican a los pocillos empleando una micropipeta 6. ánodo (+) |
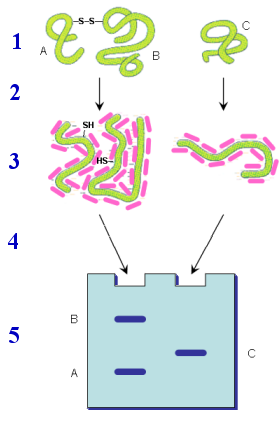 |
1. (izqda.) Proteína con dos subunidades (A y B) unidas por un puente disulfuro. 2. Se calienta con SDS y β‑mercaptoetanol 3. Moléculas de SDS (con carga negativa) recubriendo la proteína 4. Separación electroforética en gel de poliacrilamida con SDS 5. Resultado |
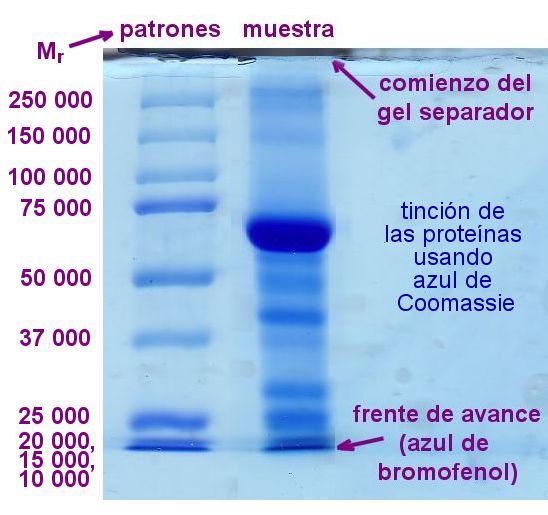
SDS-PAGE = SDS-PolyAcrylamide Gel Electrophoresis, electroforesis en gel de poliacrilamida en presencia de dodecilsulfato sódico.
Gel de poliacrilamida, en placa, vertical.
Separación de acuerdo con la masa molecular (estrictamente, con la longitud de la cadena polipeptídica).
La muestra se trata con SDS (a mayor concentración que en la composición del gel) y se calienta brevemente a 90-100°C, para provocar la desnaturalización. Se suele añadir también β‑mercaptoetanol para reducir los puentes disulfuro, separando así las subunidades de la proteína y permitiendo que se desplieguen por completo por efecto del SDS.
En la preparación del gel se mezclan:
Permite obtener una estimación de la masa molecular de las proteínas separadas. Para ello es preciso analizar en la misma electroforesis unas proteínas patrón, de tamaño conocido, con las que se construye una curva de calibrado, representando movilidad frente a logaritmo de masa molecular.
 Para controlar el avance del frente de electroforesis (posición de máxima movilidad) se añade a la muestra
un colorante marcador (“tracking dye”), molécula cargada y de pequeño tamaño que avanza más que cualquier componente de la muestra; el más habitual es el azul de bromofenol.
Para controlar el avance del frente de electroforesis (posición de máxima movilidad) se añade a la muestra
un colorante marcador (“tracking dye”), molécula cargada y de pequeño tamaño que avanza más que cualquier componente de la muestra; el más habitual es el azul de bromofenol.
Para mejorar la resolución (obteniendo bandas más compactas) se suele emplear electroforesis discontinua:
El efecto concentrador se debe a una mayor porosidad del gel acumulador combinada con una diferente composición de los tampones de cubeta (Tris-glicina) y de gel (Tris-HCl) y distinto pH para ambos geles.
Nota: “Tris” es tris(hidroximetil)aminometano, una sustancia habitual para preparar disoluciones tampón de pH próximo a 7.
| composición del gel | resolución conseguida: tamaño de DNA (kb) |
|
|---|---|---|
| % de acrilamida |
% de agarosa |
|
| 20 | 0,006 - 0,10 | |
| 15 | 0,025 - 0,15 | |
| 12 | 0,04 - 0,2 | |
| 8 | 0,06 - 0,4 | |
| 5 | 0,08 - 0,5 | |
| 3,5 | 1 - 2 | |
| 2 | 0,1 - 2 | |
| 1,5 | 0,2 - 3 | |
| 1,2 | 0,4 - 6 | |
| 0,9 | 0,5 - 7 | |
| 0,7 | 0,8 - 10 | |
| 0,6 | 1 - 20 | |
| 0,3 | 5 - 60 | |
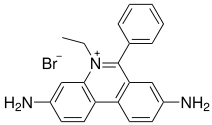
Las moléculas de DNA de las células son excesivamente grandes para avanzar a través de un gel de electroforesis normal, pero pueden analizarse si previamente se han fragmentado de forma controlada. Se suelen emplear geles de agarosa (concentración entre 0,3% y 2%), más porosos que los de poliacrilamida. Las características mecánicas de estos geles hacen aconsejable la realización de la electroforesis en horizontal, habitualmente “submarina”. Las muestras se aplican en pocillos practicados dentro del gel.
Una de las formas más comunes de fragmentar el DNA es el empleo de endonucleasas de restricción, que cortan en secuencias diana características.
La carga eléctrica de una molécula de DNA depende de sus grupos fosfato, y el número de éstos es igual al doble del número de pares de bases. La forma de todas las moléculas de DNA es esencialmente la misma. En consecuencia, resulta que la movilidad en electroforesis depende exclusivamente de la longitud de la molécula (medida habitualmente como número de pares de bases, pb): la electroforesis separa los fragmentos de DNA de acuerdo con su longitud en pb. De forma similar al caso de proteínas en SDS-PAGE, se suelen aplicar patrones en uno de los pocillos para hacer una curva de calibrado de tamaños.

1. fuente de alimentación (corriente continua entre los extremos del gel)
2. "pocillo" para depositar las muestras de DNA (fragmentado con enzimas de restricción) y los patrones
3. gel (de agarosa o poliacrilamida)
4. el gel y el tampón de separación tienen pH alcalino (el DNA adquiere carga negativa)


5. los fragmentos de DNA (carga negativa) avanzan hacia el polo positivo; a menor tamaño, mayor velocidad y por tanto mayor recorrido

6. punto de aplicación de las muestras
7. fragmentos de DNA bicatenario separados por tamaño
8. “patrones” o “marcadores” de DNA (fragmentos de tamaño conocido) (escalera de DNA)
9. distintas muestras de DNA o distintas fragmentaciones
El avance del frente de electroforesis se observa gracias a colorantes como azul de bromofenol y xileno cianol, añadidos a la muestra.
Cuando los fragmentos de ácido nucleico analizados son de tamaño muy pequeño se utilizan geles de poliacrilamida, o de mezclas de agarosa y poliacrilamida cuando el tamaño es intermedio.
 Bromuro de etidio:
Agente intercalante, se introduce entre las bases apiladas del DNA, abre un poco la doble hélice, provoca un desenrollamiento local.
Su fluorescencia (color naranja al iluminar con UV) aumenta mucho cuando se une al DNA: método de tinción o revelado del DNA, especialmente usado en electroforesis y en centrifugación.
Bromuro de etidio:
Agente intercalante, se introduce entre las bases apiladas del DNA, abre un poco la doble hélice, provoca un desenrollamiento local.
Su fluorescencia (color naranja al iluminar con UV) aumenta mucho cuando se une al DNA: método de tinción o revelado del DNA, especialmente usado en electroforesis y en centrifugación.
Se pueden emplear sustancias coloreadas o fluorescentes que se unan a las proteínas o los ácidos nucleicos.
En el primer caso suelen ser colorantes orgánicos que interaccionan con las proteínas de forma poco selectiva, principalmente electrostática. Ejemplos: azul brillante de Coomassie, negro amido, rojo Ponceau.
Para los ácidos nucleicos se usa el azul de metileno o, más frecuentemente, compuestos fluorescentes e intercalantes, como el bromuro de etidio (véase figura). Un compuesto intercalante es aquél que se introduce entre los pares de bases apilados del DNA.
Tras la electroforesis, el gel se sumerge en un recipiente con una disolución del colorante y se espera a que aquél se impregne y así el colorante se una a las moléculas separadas en el gel. Tras lavar el gel para eliminar el exceso de tinción no unida específicamente, se observa, fotografía o cuantifica mediante densitometría.
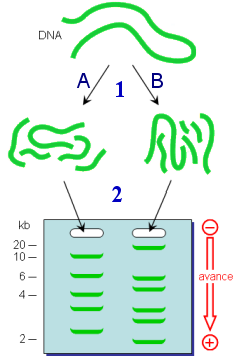
1. Se corta el DNA con la enzima de 2. Separación de los fragmentos |
 Detección mediante tinción con un agente fluorescente y observación bajo luz ultravioleta 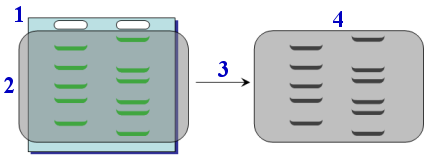 Detección mediante autorradiografía 1. gel de agarosa seco 2. película fotográfica 3. exposición y revelado de la película 4. autorradiografía revelada |
 |
Si entre las moléculas separadas hay una enzima, se puede detectar añadiendo sus sustratos (naturales o sintéticos) y sus cofactores, y detectando la aparición del producto, generalmente coloreado.
Tanto proteínas como ácidos nucleicos pueden marcarse isotópicamente previamente a la electroforesis, en cuyo caso la detección se hace mediante autorradiografía del gel (véase figura).
En caso de disponer de ellos, los anticuerpos permiten la detección selectiva del componente de interés (proteína o grupo marcador unido químicamente a la proteína o al ácido nucleico antes de la electroforesis). La permeabilidad de los geles para el acceso del anticuerpo es limitada, por lo que se hace una transferencia de las moléculas separadas en el gel hacia una membrana de nitrocelulosa o de nailon, la cual se somete luego a la disolución que contiene el anticuerpo. Los detalles de esta técnica de inmunotransferencia o ensayo Western blot se tratarán en un tema posterior.
Las moléculas con una secuencia determinada de nucleótidos se pueden detectar mediante hibridación con sondas específicas, pequeñas moléculas de ácido nucleico monocatenario (oligonucleótidos) con la secuencia complementaria a la buscada y marcadas con un isótopo radiactivo, un grupo fluorescente, etc. Para llevar a cabo con éxito la hibridación se requiere también la transferencia desde el gel a una membrana de nitrocelulosa o nailon. Los detalles de estos ensayos (Southern blot para DNA y Northern blot para RNA) se estudian en un tema posterior.
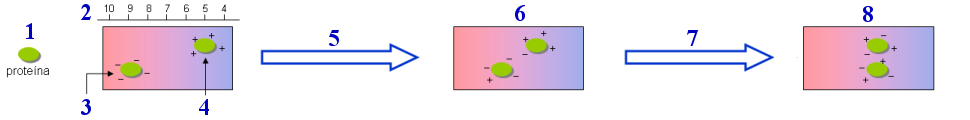
(También se llama enfoque isoeléctrico o IEF, de isoelectrofocusing). Se trata de una variante de electroforesis en la que el gel (poliacrilamida) posee un gradiente de pH fijado en su estructura. Esto se consigue incluyendo en su preparación moléculas ionizables con valores de pK diferentes, enlazadas de forma covalente al polímero que forma el gel. Como consecuencia, al avanzar los componentes de la muestra a lo largo del gel se van encontrando con entornos de pH diferente, y eventualmente alcanzan una región en la cual el pH local es igual al punto isoeléctrico de la molécula y, en consecuencia, ésta detiene su avance. Se alcanza, pues, un equilibrio y se consigue la separación de los componentes de la muestra de acuerdo con su punto isoeléctrico, que además se puede medir, pues se conoce la geometría del gradiente de pH fijado al gel.
1. Proteína con punto isoeléctrico de 6.5 (pI=6.5) 2. Gradiente de pH inmovilizado |
5. Su carga hace que las moléculas de proteína se desplacen en el campo eléctrico |
7. La carga hace que las moléculas de proteína sigan desplazándose |
||
 |
||||
3. En un entorno de pH > pI, la proteína tiene carga negativa 4. En un entorno de pH < pI, la misma proteína tiene carga positiva |
6. Al moverse, cambia el pH de su entorno, con lo que la proteína va perdiendo carga neta |
8. Al alcanzar un entorno con pH = pI, la proteína ya no tiene carga neta y no se mueve más |
||
Tras realizar una primera separación, su resultado se somete en dirección perpendicular a otra de diferente mecanismo. Generalmente la primera es un isoelectroenfoque en un gel cilíndrico estrecho que luego se coloca como muestra para una SDS-PAGE. Se obtienen así mapas complejos de bandas, muy característicos de cada muestra, cuya utilidad principal es la comparación con el obtenido de otra muestra similar pero conocida.


Las moléculas o fragmentos de DNA de longitud superior a 20-40 kb no se pueden resolver (separar) empleando la electroforesis convencional en gel de agarosa. Esto se debe, en primer lugar, a la dificultad de manejo de los geles preparados con las bajas concentraciones de agarosa que requerirían (inferior al 0,4%). Además, las moléculas de DNA, que adoptan una conformación más o menos globular, poseen un tamaño excesivamente grande para atravesar los poros del gel y no se separan en función de su tamaño. Para solventar este problema se ha ideado una modificación de la técnica, conocida como electroforesis de campo pulsante (o pulsado, pulsed-field gel electrophoresis, PFGE). Se emplean geles de agarosa al 1%, pero se altera periódicamente la orientación del campo eléctrico aplicado, activando alternativamente dos pares de electrodos. El frecuente cambio de dirección del campo eléctrico consigue que las moléculas se desplieguen y avancen a través de los poros en conformación extendida. A mayor tamaño, se reorientan con más dificultad, por lo que avanzan más despacio.

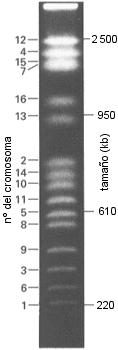 Así se consigue separar fragmentos de DNA de hasta varias megabases, lo que supone un avance significativo pero aún no
sirve para estudiar los cromosomas humanos enteros (de 50 a 250 Mb cada uno). Sí se han podido analizar así los cromosomas de levadura (figura derecha).
Así se consigue separar fragmentos de DNA de hasta varias megabases, lo que supone un avance significativo pero aún no
sirve para estudiar los cromosomas humanos enteros (de 50 a 250 Mb cada uno). Sí se han podido analizar así los cromosomas de levadura (figura derecha).
Las muestras han de contener el DNA sin fragmentar, por lo se requiere una preparación especial (al purificar el DNA por los métodos habituales se fragmenta por debajo de 100 kb). Para obtener preparaciones con los cromosomas intactos se mezclan las células con agarosa caliente (37°C) y se vierte en pequeños moldes de unos milímetros, de forma que al solidificarse la agarosa (4°C) forma bloques (“insertos”) que incluyen las células intactas. En éstos se realiza la lisis celular y la digestión de las proteínas (p.ej., con EDTA, SDS y proteinasa K, que difunden a través de los poros del gel) sin que se alteren las grandes moléculas de DNA. Tras una diálisis para eliminar los restos de la digestión, se consigue un bloque de agarosa con el DNA intacto en su interior, y se deposita el bloque entero en un pocillo del gel de electroforesis.
Se han desarrollado variantes preparativas, es decir, que permiten la obtención de cantidades significativas de los componentes de la muestra por separado. No se detallarán aquí (véase Freifelder 1991, pp.256-7)
Combina una separación electroforética con una detección por inmunodifusión.
Como se ha indicado, se puede obtener una estimación de la masa molecular (en kDa) de proteínas empleando SDS-PAGE, y de fragmentos de DNA (en pb) empleando electroforesis en agarosa. En ambos casos, en uno de los pocillos del gel se carga una mezcla de moléculas patrón de tamaño conocido, con el fin de poder trazar la curva de calibrado. Para las proteínas, la validez del dato obtenido depende de que la desnaturalización con SDS haya sido completa y de que se hayan separado las subunidades gracias a la reducción con mercaptoetanol; además, la presencia de oligosacáridos en las glicoproteínas altera la movilidad, que ya no proporciona valores de masa molecular fiables.
Ejemplo 1: Se aplica en uno de los pocillos del gel una mezcla de fragmentos de DNA de tamaño conocido, denominados “patrones”, “marcadores de masa molecular” o “escalera de DNA” (DNA ladder). Tras su separación en la electroforesis, se revelan y se representa para todas las bandas la movilidad (distancia avanzada o, mejor, cociente entre ésta y el avance del frente de electroforesis) frente al logaritmo del tamaño (longitud en pb). Sobre la línea de regresión se interpolan las distancias de avance de las muestras problema, calculando así su tamaño en pb. En este ejemplo, los patrones empleados son fragmentos bien conocidos, aquellos obtenidos a partir del DNA del virus λ por la acción de las endonucleasas de restricción Eco RI e Hin dIII. |
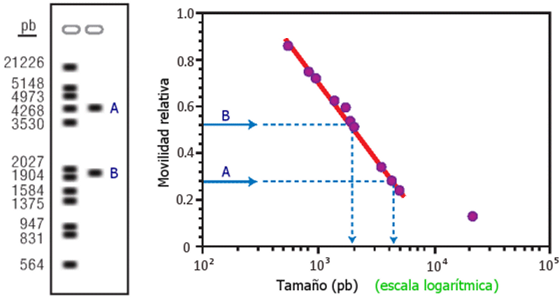 |
 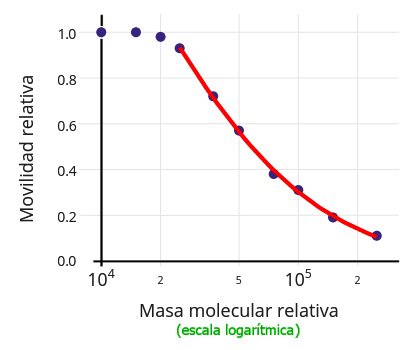 |
Ejemplo 2: Se aplica en uno de los pocillos de un gel SDS-PAGE una mezcla de proteínas de una sola subunidad, de tamaño conocido, denominadas “patrones” o “marcadores de masa molecular”. Tras su separación en la electroforesis, se revelan y se representa para todas las bandas la movilidad (distancia avanzada o, mejor, cociente entre ésta y el avance del frente de electroforesis) frente al logaritmo del tamaño (masa molecular relativa, Mr). Sobre la línea de regresión se interpolan las distancias de avance de las muestras problema, calculando así su masa molecular aparente. |
Como se ha indicado, mediante isoelectroenfoque [↑] se obtiene una medida del punto isoeléctrico de las proteínas (pHI o pI).
Las variantes de una enzima, con pequeñas diferencias en su estructura y en su actividad enzimática, se analizan rutinariamente mediante electroforesis o, en especial, mediante isoelectroenfoque. De hecho, los nombres de las isoenzimas derivan a veces de su movilidad electroforética. Se puede aplicar análogamente a las isoformas de proteínas no enzimáticas.
Una de las formas de estudiar la secuencia de los ácidos nucleicos, en especial el DNA, consiste en tratarlo con distintas enzimas de restricción, analizar mediante electroforesis en gel de agarosa los fragmentos resultantes e intentar reconstruir la secuencia de la molécula original formando su mapa de restricción, uno de los tipos de mapa físico.
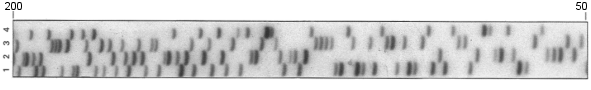
La obtención de la secuencia completa, nucleótido a nucleótido, también depende del análisis electroforético de fragmentos de DNA, tanto en el método químico de Maxam y Gilbert como en el método enzimático de Sanger. Se utilizan en este caso geles de poliacrilamida, debido al tamaño pequeño de los fragmentos, pudiéndose resolver fragmentos que se diferencian en un solo nucleótido.
| longitud de la molécula (nº de nucleótidos o pares de bases, pb) | |
| muestras (calles o carriles del gel) |
 |