 activar el proceso (marcando la casilla
);
activar el proceso (marcando la casilla
); La optimización estructural realizada en Jmol no respeta la estereoquímica de los modelos. Para intentar paliar esta limitación, se ofrecen unas “opciones avanzadas”, aunque no hay garantía de que lo consigan.
Aplica una operación de simetría de reflexión a todo el modelo 3D, generando así la forma enantiómera.
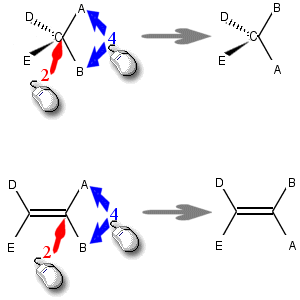
Abre un diálogo para realizar la operación de forma semimanual sobre el modelo 3D. Se aplicará una operación de intercambio de posiciones de dos de los sustituyentes de un átomo de carbono dado (esto sólo funciona cuando dicho átomo tiene 4 sustituyentes, o cuando tiene 3 si uno de ellos está unido mediante doble enlace). Se puede así generar el diastereómero en un C quiral o interconvertir los isómeros cis y trans.
La secuencia de acciones requeridas (guiada mediante indicaciones en el panel de Jmol) es:
activar el proceso (marcando la casilla
); Se puede cancelar el proceso quitando la marca a la casilla “activar”.
Al terminar convendrá aplicar una nueva optimización para ajustar la nueva estructura.
Mientras esté marcada esta casilla, se puede pulsar con el puntero del ratón sobre un átomo y arrastrarlo a otra posición en el espacio. Los átomos de hidrógeno unidos a él se moverán también. Igualmente es posible mover un átomo de hidrógeno.
Si la segunda casilla está marcada, al soltar el botón del ratón se optimizará la estructura hacia una conformación razonable. Si no lo está, se pueden mover sucesivamente varios átomos y luego pulsar el botón Optimizar.
Este procedimiento puede servir para conseguir conformaciones diferentes de la obtenida en la optimización inicial. Por ejemplo, para cambiar la orientación arriba/abajo de los sustituyentes en un anillo, intercambiar isómeros cis-trans o cambiar la estereoquímica en torno a un átomo.