Colors used by Jmol
Este documento en español
Objects can be custom colored in Jmol using the color (or colour) command:
color object color or color scheme
- color is specified as decimal [red,green,blue] or hexadecimal [xRRGGBB] triplets.
It can also be a JavaScript color name, a RasMol color name, or a Jmol color name. (For the default colors and color values used in Jmol, see table below.)
- color scheme is specified using pre-defined terms detailed on this page.
- object can be atoms, bonds, backbone, cartoon, stars, rockets, ribbon, dots, label, echo, hbonds, ssbonds, axes, boundbox, measure, polyhedra, isosurface, pmesh, unitcell...
- if the object is omitted, atoms is assumed
Information on RasMol colors is included for those who are adapting RasMol and Chime-based materials for use in Jmol.
Many of the following coloring patterns apply only to PDB and mmCIF files for biomacromolecules.
Related commands:
color object cpk , set defaultColors Jmol , set defaultColors Rasmol
Applies color to each atom of the object according to element, as shown in the tables below. Backbone displays (such as ribbons, cartoons, etc.) are rendered in the color of alpha carbons for proteins, phosphorus for nucleic acids.
Default element colors, by periodic table:
Hover over any element to see more information.
Default element colors, by atomic number:
“CPKnew” scheme applies only to Rasmol v. 2.7.3 (or later); only differences with classic CPK are shown.
Note: CPKnew for unknown atoms is FA1691,
slightly different from CPK FF1493,
but has been omitted for clarity in the table.
Related commands: color object amino , color object shapely
amino renders each of the 20 standard amino acid residues (as well as Asx and Glx)
in a certain color, along with one additional color for anything else (including nucleotides, solvents, and non-amino ligands).
Some colors are shared by two or more amino acids with similar properties.
shapely uses a different set of colors for amino acids (each one different) and also colors differentially the 6 kinds of nucleotides.
Related command: color object chain
Renders each chain in the structure in a different color. This color scheme is particularly useful for distinguishing the parts of a multimeric structure or the individual DNA strands of a double helix.
Atoms in HETERO groups (PDB files only) are colored darker than those in ATOM records.
Related command: color object structure
Uses different colors to distinguish six types of protein secondary structures (three types of helices, beta strands or sheets, turns, and loops) and DNA vs. RNA. The secondary structure is either read from the PDB file (HELIX and SHEET records), if available, or determined using an algorithm.
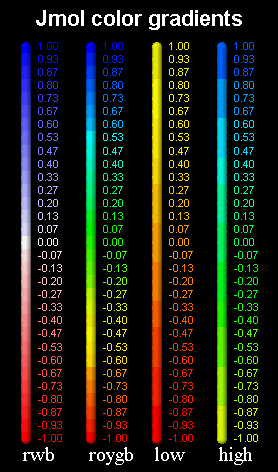
Gradients
Several properties that adopt a continuous range of values can be used to color atoms and structures. In these cases, several color gradients or "rainbows" are used by Jmol.
Direct rainbow (roygb)
red → orange → yellow → green → blue
Reverse rainbow (bgyor)
blue → green → yellow → orange → red
Red-white-blue gradient (rwb)
red → tints of red-pink → white → tints of blue → blue
Blue-white-red gradient (bwr)
blue → tints of blue → white → tints of pink-red → red
Low rainbow
red → orange → yellow
High rainbow
yellow → green → blue
| 
|
Position along chain
Related commands: color object group , color object monomer
A reverse rainbow gradient (bgyor) is used to color according to position of the corresponding groups or residues (for example, amino acids or nucleotides) along a chain.
(blue = nitrogen = N-terminal = start of chain = 5')
(red = oxygen = C-terminal = end of chain = 3')
group:
- The full color range is applied across the specified or selected groups in the chain (that is, the scale is relative, not absolute).
- The order of groups in the file is used, not the number assigned to them in the file.
- The coloring operates independently for each chain.
monomer:
A variation, relating to "monomers" in a "polymer" rather than "groups" in a "chain". A "polymer" is a series of connected "monomers" (at least for these purposes).
- Jmol interprets the backbone pattern of consecutive residues ("monomers") to identify continuous "polymers".
- If a chain has a break in it, the 2 pieces of the chain are recognized as separate polymers, and hence each one is colored independently with the full color range, despite the fact that both parts are designated with the same chain ID in the coordinate file.
- Again, the numbering of groups in the file is irrelevant.
- HETERO groups may participate in a polymer, depending upon how the PDB/mmCIF file is marked up, and as long as they have a backbone pattern that Jmol can recognize as a sequence of individual units.
RasMol/Chime note: In RasMol/Chime, group index is relative to the full chain, while in Jmol it is relative to the currently selected groups of the chain. In addition, the set hetero command is not implemented in Jmol. If you want to exclude "hetero" groups, then do not select them.
Related commands: color object relativeTemperature , color object fixedTemperature
Colors each atom, using a bwr gradient, according to the "temperature factor" (B factor, or Debye-Waller factor) value stored in the PDB or mmCIF file. Typically this gives a measure of the mobility or uncertainty of a given atom's position. A high crystallographic B factor may indicate an incorrect structure.
(blue = cold = low temperature = static/immobile)
(red = hot = high temperature = variable/mobile)
fixedTemperature: the factor is made relative to an absolute scale of 0 to 100.
relativeTemperature: the color is relative to the lowest and highest B factor values within the file (not within the selection).
relativeTemperature after set rangeSelected on: the color is relative to the lowest and highest B factor values within the currently selected part of the model.
Note: The temperature factor fields in a PDB file are sometimes used by the file creator to indicate any other characteristic of the atoms in the model.
RasMol/Chime note: In RasMol and Chime, temperature (the only option available) uses also a relative scale, but with a reverse rainbow gradient (bgyor). In Jmol, temperature does the same as relativeTemperature.
Formal charge
Related command: color object formalCharge
Colors atoms based on their formal charge, or ionic state. Uses a restricted rwb gradient: tints of red for negative, tints of blue for positive, white for uncharged atoms. The range of values handled by Jmol for formal charge is -4 through +7, in an absolute scale.
(red = oxygen = carboxy = negative)
(blue = nitrogen = amino = positive)
Partial charge
Related command: color object partialCharge
Colors atoms based on their partial charge, or electron density. Uses a rwb gradient: tints of red for high electron density, tints of blue for low electron density, white for neutral. The range of values for partial charge is adjusted to the minimum and maximum values present in the file (i.e., a scale relative to the file values is used, but not to values in the selected set of atoms).
(red = oxygen = carboxy = negative)
(blue = nitrogen = amino = positive)
RasMol/Chime note: In RasMol and Chime, charge (the only option available) is meant for PDB files that hold partial charges in the B factor columns, and uses a direct rainbow gradient (roygb) with a scale relative to values present in file. In Jmol, charge does the same as formalCharge.
Related command: isoSurface
Isosurfaces (solvent, molecular, orbitals...) can be colored uniformly or using a color map that reflects the value of some property at each point in the surface (such as molecular electrostatic potential, distance, etc.). For these maps all the predefined gradients of color are available (and the reverse gradients too), using the colorScheme parameter of isoSurface command.
Related command: color hBonds type
Colors hydrogen bonds in a protein based on the number of residues between the atoms participating in the bond. A different color is used for hydrogen bonds in nucleic acids.